|
|
Pathology definition - Thalassemia
Thalassemia
There are two different form of thalassemia. These include alpha thalassemia and beta thalassemia. Both thalassemia mostly affecting people of Mediterranean ancestry.
Alpha thalassemia consists of four different types. The first variant is alpha thalassemia trait which is asymptomatic and present with 3 -4 normal gene. The second variant is alpha thalassemia trait with the present of 2 normal genes. There will be a decreased in the MCV which lead to mild anemia. The third variant is hemoglobin H disease with the present of 1 normal gene and consists of Hgb H due to aggregation of the excess of the beta chain, decreased MCV, severe hemolytic anemia an d splenomegaly. The fourth variant is hydrops fetalis with no normal gene and commonly associated with stillborn.
Alpha thalassemia may present with target cells, hypochromic microcytic erythrocytes in the peripheral blood smear. Alpha thalassemia is not treated for the alpha thalassemia trait. However, transfusion is considered in cases of hemoglobin H disease and alpha thalassemia minor.
Alpha thalassemia commonly occur due to deletion of one or more four gene which coded for the alpha globin chain of the Hgb.
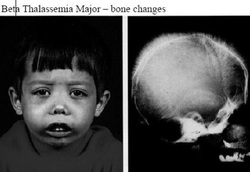
Beta thalassemia consists of only two variant which include beta thalassemia minor and beta thalassemia major. Beta thalassemia minor is a heterozygosity disorder that present with decreased in the MCV and mild anemia. Beta thalassemia major is a homozygosity disorder that present with decreased MCV and severe hemolytic anemia m splenomegaly, hemosiderosis, heart failure due to hemosiderosis,increased in Hgb F and abnormalities of the long bone and facial bone. Beta thalassemia major commonly present in infancy.
Beta thalassemia may present with anisopoikilocytosis , target cells and hypochromic microcytic erythrocytes. Beta thalassemia may require bone marrow transplantation and transfusion for beta thalassemia major.However no treatment is required for beta thalassemia major.
Beta thalassemia may occur due to point mutation of the beta globin gene. This will result in reduction or absent in the synthesis of the beta globin chain of the Hgb.
References
1.Weatherall, David. “Thalassemias.” In eLS. John Wiley & Sons, Ltd, 2001.
2.Sonakul, D, P Pacharee, and K Thakerngpol. “Pathologic Findings in 76 Autopsy Cases of Thalassemia.” Birth Defects Original Article Series 23, no. 5B (1988): 157–176.
There are two different form of thalassemia. These include alpha thalassemia and beta thalassemia. Both thalassemia mostly affecting people of Mediterranean ancestry.
Alpha thalassemia consists of four different types. The first variant is alpha thalassemia trait which is asymptomatic and present with 3 -4 normal gene. The second variant is alpha thalassemia trait with the present of 2 normal genes. There will be a decreased in the MCV which lead to mild anemia. The third variant is hemoglobin H disease with the present of 1 normal gene and consists of Hgb H due to aggregation of the excess of the beta chain, decreased MCV, severe hemolytic anemia an d splenomegaly. The fourth variant is hydrops fetalis with no normal gene and commonly associated with stillborn.
Alpha thalassemia may present with target cells, hypochromic microcytic erythrocytes in the peripheral blood smear. Alpha thalassemia is not treated for the alpha thalassemia trait. However, transfusion is considered in cases of hemoglobin H disease and alpha thalassemia minor.
Alpha thalassemia commonly occur due to deletion of one or more four gene which coded for the alpha globin chain of the Hgb.
Beta thalassemia consists of only two variant which include beta thalassemia minor and beta thalassemia major. Beta thalassemia minor is a heterozygosity disorder that present with decreased in the MCV and mild anemia. Beta thalassemia major is a homozygosity disorder that present with decreased MCV and severe hemolytic anemia m splenomegaly, hemosiderosis, heart failure due to hemosiderosis,increased in Hgb F and abnormalities of the long bone and facial bone. Beta thalassemia major commonly present in infancy.
Beta thalassemia may present with anisopoikilocytosis , target cells and hypochromic microcytic erythrocytes. Beta thalassemia may require bone marrow transplantation and transfusion for beta thalassemia major.However no treatment is required for beta thalassemia major.
Beta thalassemia may occur due to point mutation of the beta globin gene. This will result in reduction or absent in the synthesis of the beta globin chain of the Hgb.
References
1.Weatherall, David. “Thalassemias.” In eLS. John Wiley & Sons, Ltd, 2001.
2.Sonakul, D, P Pacharee, and K Thakerngpol. “Pathologic Findings in 76 Autopsy Cases of Thalassemia.” Birth Defects Original Article Series 23, no. 5B (1988): 157–176.
