|
|
Pathology definition - Sickle cell disease
Sickle cell disease
Sickle cell disease or sickle cell anemia is caused by mutation of the gene coding for the production of the beta globin chain of the Hgb. The mutation is associated with the replacement of the valine for glutamine). This is an autosomal recessive disorder which lead to the production of the Hgb S. Sickle cell trait is associated with heterozygous for the Hgb S gene)
Hemoglobin C disease is associated with another variant in the mutation of the beta chain of the Hgb.
Sickle cell disease/sickle cell anemia is associated with vaso - occlusive crisis that is characterized by pain in the back and the limbs which is severe in nature due to the blockage of the microvasculature by sickled cell. Sickle cell disease is also presented with signs and symptoms such as leg ulcers, jaundice, chronic hemolytic anemia, repeated infarction ( autosplenectomy) and increase susceptibility to the infection by encapsulated organism. Besidesa that sickle cell disease, may also lead to aplastic crises due to provocation by B19 parvovirus infection.
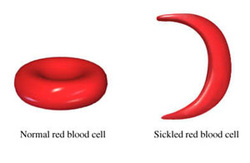
Sickle cell disease/sickle cell anemia is commonly characterized by reticulocytosis, the present of Howell - Jolly bodies and cresent shaped red blood cell on the peripheral blood smear. In hypoxic environment, Hgb S will polymerizes which lead to distortion of the shape of the red blood cell and increase the susceptibility to hemolysis. Hgb S may polymerized due to dehydration, extreme work out or infection. However, The hgb S may provide protection against P. falciparum malaria.
The treatment for sickle cell disease /sickle cell anemia may include hydroxyurea and transfusion in cases of vaso occlusive crises or hemolytic condition. Hydroxyurea may lead to an increase in the Hgb F levels.
References
1.Rees, David C, Thomas N Williams, and Mark T Gladwin. “Sickle-Cell Disease.” The Lancet 376, no. 9757 (December 2010): 2018–2031. doi:10.1016/S0140-6736(10)61029-X.
2.MASON VR. “SIckle Cell Anemia.” Journal of the American Medical Association 79, no. 16 (October 14, 1922): 1318–1320. doi:10.1001/jama.1922.02640160038012.
Sickle cell disease or sickle cell anemia is caused by mutation of the gene coding for the production of the beta globin chain of the Hgb. The mutation is associated with the replacement of the valine for glutamine). This is an autosomal recessive disorder which lead to the production of the Hgb S. Sickle cell trait is associated with heterozygous for the Hgb S gene)
Hemoglobin C disease is associated with another variant in the mutation of the beta chain of the Hgb.
Sickle cell disease/sickle cell anemia is associated with vaso - occlusive crisis that is characterized by pain in the back and the limbs which is severe in nature due to the blockage of the microvasculature by sickled cell. Sickle cell disease is also presented with signs and symptoms such as leg ulcers, jaundice, chronic hemolytic anemia, repeated infarction ( autosplenectomy) and increase susceptibility to the infection by encapsulated organism. Besidesa that sickle cell disease, may also lead to aplastic crises due to provocation by B19 parvovirus infection.
Sickle cell disease/sickle cell anemia is commonly characterized by reticulocytosis, the present of Howell - Jolly bodies and cresent shaped red blood cell on the peripheral blood smear. In hypoxic environment, Hgb S will polymerizes which lead to distortion of the shape of the red blood cell and increase the susceptibility to hemolysis. Hgb S may polymerized due to dehydration, extreme work out or infection. However, The hgb S may provide protection against P. falciparum malaria.
The treatment for sickle cell disease /sickle cell anemia may include hydroxyurea and transfusion in cases of vaso occlusive crises or hemolytic condition. Hydroxyurea may lead to an increase in the Hgb F levels.
References
1.Rees, David C, Thomas N Williams, and Mark T Gladwin. “Sickle-Cell Disease.” The Lancet 376, no. 9757 (December 2010): 2018–2031. doi:10.1016/S0140-6736(10)61029-X.
2.MASON VR. “SIckle Cell Anemia.” Journal of the American Medical Association 79, no. 16 (October 14, 1922): 1318–1320. doi:10.1001/jama.1922.02640160038012.
