|
|
Pathology definition - Jaundice
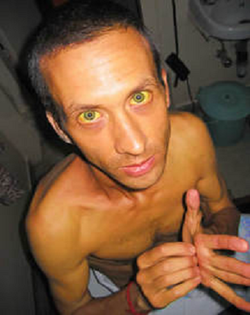
Jaundice
Jaundice is yellow discoloration of the sclera, skin and tissue.
Pathologically jaundice is caused by increase production of bilirubin, impairment of the flow of bilirubin , accumulation of excess bilirubin and deficiency in conjugation.
Normally, the whole process begin with heme. Heme is later degraded into bilirubin. Heme degradation may occur outside the liver. The bilirubin is then bound to albumin and finally transported to the liver/hepatocytes system for further processing. In liver, the bilirubin will undergoes conjugation process and this newly form conjugated bilirubin will be excreted into a bile. The bile will enter the gastrointestinal tract and with the help of intestinal bacteria converted into urobilinogen and reabsorbed from the small intestine and excreted in the urine.
The causes of jaundice are hepatocellular disease, hemolytic anemia and biliary obstruction as well as congenital hyperbilirubinemia. Hepatocellular disease is presented with the raised ALT, AST and ALP ( liver enzyme), increase in urine bilirubin and decreased or normal urobilinogen. Hepatocellular jaundice is associated with unconjugated and conjugated hyperbilirubinemia.
Hemolytic anemia is associated with unconjugated hyperbilirubinemia. The patient may present with increase level of the urine urobilinogen and acholuria/absent of urine bilirubin
Biliary obstruction may present with conjugated hyperbilirubinemia with hypercholesterolemia, decrease urine urobilinogen and raised liver enzyme ALP as well as increase in urine bilirubin.
Congenital hyperbilirubinemia consists of Dubin - Johnson syndrome and Rotor syndrome, Gilbert syndrome and Type 1 and type 2 Crigler- Najjar syndrome.
Dubin - Johnson syndrome and Rotor syndrome are autosomal recessive condition . Patient typically present with epigastric and right upper quadrant pain as well as jaundice. Dubin - Johnson syndrome is mostly associate with black liver, slightly elevated liver enzyme and conjugated hyperbilirubinemia. Dubin Johnson syndrome and Rotor syndrome typically affecting the transport of bilirubin out of the liver.
Gilbert syndrome is an autosomal dominant condition. Patient may present with unconjugated hyperbilirubinemia and mild scleral icterus. The patient may remain asymptomatic and the condition may be triggered by stress. Gilbert syndrome results in decreased UDP - glucuronyl trasnferase activity which decrease bilirubin uptake by the liver.
Type 1 Crigler Najjar syndrome is an autosomal recessive condition while type 2 Crigler Najjar syndrome is an autosomal dominant condition with variable penetrance. Patient may present with unconjugated hyperbilirubinemia and kernicterus. Type 1 Crigler Najjar syndrome appears to be more severe than type 2 Crigler Najjar syndrome. Type 1 and Type2 Crigler Najjar syndrome are may lead to absent of the UDP - glucuronyl transferase activity. The treatment of Type 1 Crigler Najjar syndrome consists of phototherapy and plasmapheresis while type 2 Criggler Najjar syndrome is treated with phenobarbital.
Neonatal hyperbilirubinemia may occur during the first few week of life due to immature liver which lead to deficiency of the UDP - glucuronyl transferase and increase production of bilirubin.
References
1.Malchow-Møller, A, P Matzen, B Bjerregaard, J Hilden, J Holst-Christensen, T Staehr Johansen, L Altman, C Thomsen, and E Juhl. “Causes and Characteristics of 500 Consecutive Cases of Jaundice.” Scandinavian Journal of Gastroenterology 16, no. 1 (1981): 1–6.
2.Nisa, Aziz-un, and Zubair Ahmad. “Dubin-Johnson Syndrome.” Journal of the College of Physicians and Surgeons Pakistan (March 1, 2008): 188–9.
3.Kaplan, Michael, Paul Renbaum, Ephrat Levy-Lahad, Cathy Hammerman, Amnon Lahad, and Ernest Beutler. “Gilbert Syndrome and Glucose-6-phosphate Dehydrogenase Deficiency: A Dose-dependent Genetic Interaction Crucial to Neonatal Hyperbilirubinemia.” Proceedings of the National Academy of Sciences 94, no. 22 (October 28, 1997): 12128–12132.
Jaundice is yellow discoloration of the sclera, skin and tissue.
Pathologically jaundice is caused by increase production of bilirubin, impairment of the flow of bilirubin , accumulation of excess bilirubin and deficiency in conjugation.
Normally, the whole process begin with heme. Heme is later degraded into bilirubin. Heme degradation may occur outside the liver. The bilirubin is then bound to albumin and finally transported to the liver/hepatocytes system for further processing. In liver, the bilirubin will undergoes conjugation process and this newly form conjugated bilirubin will be excreted into a bile. The bile will enter the gastrointestinal tract and with the help of intestinal bacteria converted into urobilinogen and reabsorbed from the small intestine and excreted in the urine.
The causes of jaundice are hepatocellular disease, hemolytic anemia and biliary obstruction as well as congenital hyperbilirubinemia. Hepatocellular disease is presented with the raised ALT, AST and ALP ( liver enzyme), increase in urine bilirubin and decreased or normal urobilinogen. Hepatocellular jaundice is associated with unconjugated and conjugated hyperbilirubinemia.
Hemolytic anemia is associated with unconjugated hyperbilirubinemia. The patient may present with increase level of the urine urobilinogen and acholuria/absent of urine bilirubin
Biliary obstruction may present with conjugated hyperbilirubinemia with hypercholesterolemia, decrease urine urobilinogen and raised liver enzyme ALP as well as increase in urine bilirubin.
Congenital hyperbilirubinemia consists of Dubin - Johnson syndrome and Rotor syndrome, Gilbert syndrome and Type 1 and type 2 Crigler- Najjar syndrome.
Dubin - Johnson syndrome and Rotor syndrome are autosomal recessive condition . Patient typically present with epigastric and right upper quadrant pain as well as jaundice. Dubin - Johnson syndrome is mostly associate with black liver, slightly elevated liver enzyme and conjugated hyperbilirubinemia. Dubin Johnson syndrome and Rotor syndrome typically affecting the transport of bilirubin out of the liver.
Gilbert syndrome is an autosomal dominant condition. Patient may present with unconjugated hyperbilirubinemia and mild scleral icterus. The patient may remain asymptomatic and the condition may be triggered by stress. Gilbert syndrome results in decreased UDP - glucuronyl trasnferase activity which decrease bilirubin uptake by the liver.
Type 1 Crigler Najjar syndrome is an autosomal recessive condition while type 2 Crigler Najjar syndrome is an autosomal dominant condition with variable penetrance. Patient may present with unconjugated hyperbilirubinemia and kernicterus. Type 1 Crigler Najjar syndrome appears to be more severe than type 2 Crigler Najjar syndrome. Type 1 and Type2 Crigler Najjar syndrome are may lead to absent of the UDP - glucuronyl transferase activity. The treatment of Type 1 Crigler Najjar syndrome consists of phototherapy and plasmapheresis while type 2 Criggler Najjar syndrome is treated with phenobarbital.
Neonatal hyperbilirubinemia may occur during the first few week of life due to immature liver which lead to deficiency of the UDP - glucuronyl transferase and increase production of bilirubin.
References
1.Malchow-Møller, A, P Matzen, B Bjerregaard, J Hilden, J Holst-Christensen, T Staehr Johansen, L Altman, C Thomsen, and E Juhl. “Causes and Characteristics of 500 Consecutive Cases of Jaundice.” Scandinavian Journal of Gastroenterology 16, no. 1 (1981): 1–6.
2.Nisa, Aziz-un, and Zubair Ahmad. “Dubin-Johnson Syndrome.” Journal of the College of Physicians and Surgeons Pakistan (March 1, 2008): 188–9.
3.Kaplan, Michael, Paul Renbaum, Ephrat Levy-Lahad, Cathy Hammerman, Amnon Lahad, and Ernest Beutler. “Gilbert Syndrome and Glucose-6-phosphate Dehydrogenase Deficiency: A Dose-dependent Genetic Interaction Crucial to Neonatal Hyperbilirubinemia.” Proceedings of the National Academy of Sciences 94, no. 22 (October 28, 1997): 12128–12132.
